info@annprecisionmed.org

Case Report Volume 1, Issue 1
Importance of using new and multiple interpretive tools and algorithms in genetic variants with extremely low population frequency
Ana Katerin Minota Idárraga1,3*; Lina Johanna Moreno Giraldo2-4
1Pediatric Resident, University Libre, Cali Section, Cali, Colombia.
2Medical Genetics, Postgraduate in Clinical Bioinformatics and Genomics, Postgraduate Professor at the Health Faculty, University Libre, Cali Section, Cali, Colombia.
3Pediatric Research Group (GRINPED), Cali, Colombia.
4Neurogenetic and Metabolic Diseases Research Line, Cali, Colombia
Corresponding Author: Ana Katerin Minota Idarraga
Email ID: anakaterineminota1017@gmail.com
Copyright: © Minota AKI (2024). This Article is distributed under the terms of Creative Commons Attribution 4.0 International License
Annals of Precision Medicine
Received: Nov 14, 2024
Accepted: Dec 16, 2024
Published Online: Dec 23, 2024
Citation: Minota AKI, Moreno LJG. Importance of using new and multiple interpretive tools and algorithms in genetic variants with extremely low population frequency. Ann Precis Med. 2024; 1(1): 1002.
Abstract
Introduction: Familial hypercholesterolemia (FH) is an autosomal dominant genetic disorder presenting in heterozygous (HeFH) and homozygous (HoFH) forms. Pathogenic variants in the APOB gene are associated with FH type 2 or HoFH, which may result in early cardiovascular manifestations. The identification and characterization of novel genetic variants, combined with techniques such as reverse phenotyping and integrative interpretative algorithms, are essential for early diagnosis, appropriate management, follow-up, and prevention of cardiovascular complications.
Clinical case: We report a 2-year-4-month-old female with persistently elevated LDL levels since 6 months of age, refractory to nutritional modifications and omega-3 fatty acid supplementation. The patient was born at term with an uncomplicated delivery. Family history was significant for a maternal grandmother with hypercholesterolemia and hepatic steatosis, and a maternal great-grandmother with cerebrovascular disease, with no other known genetic lipid disorders. Physical examination revealed acanthosis nigricans and mild hepatomegaly. Given the high clinical suspicion for a genetic lipid metabolism disorder, molecular analysis was performed using whole-exome sequencing (WES) and copy number variation (CNV) analysis.
Discussion: Molecular testing identified a heterozygous variant in the APOB gene (c.7681C>T, p.Leu2561Phe), initially classified as a variant of uncertain significance (VUS). This novel variant was not previously reported in major databases including NCBI, MedGen, HGMD, OMIM, 1000 Genomes Project, ExAC, LOVD3, Ensembl, ClinGen, or DGV. In silico prediction algorithms subsequently modified its classification from benign to uncertain. Following the updated American College of Medical Genetics and Genomics criteria (2024), it was reclassified as likely pathogenic. Comprehensive analysis using biological databases, genomic annotations, molecular modeling, protein structure-function analysis, and artificial intelligence platforms predicted a deleterious effect, enabling genotype-phenotype correlation through reverse phenotyping. Recent literature has emphasized the significance of genetic testing in FH, particularly highlighting the heterogeneity of APOB variants and the importance of genotype-phenotype correlations.
Conclusion: This case underscores the significance of cascade screening and genetic counseling for family members. The implementation of advanced methodologies, including artificial intelligence and in silico prediction tools, facilitates the reclassification of uncertain variants, thereby supporting evidence-based clinical decision-making. Furthermore, early molecular diagnosis enables targeted therapeutic interventions, appropriate genetic counseling, segregation studies, clinical monitoring, and prognostication, advancing the field toward precision medicine: personalized, predictive, preventive, participatory, and proactive, with potential population-level applications (7P medicine).
Introduction
Familial Hypercholesterolemia (FH) is a rare autosomal dominant disorder that occurs in two forms: Heterozygous Familial Hypercholesterolemia (HeFH) and Homozygous Familial Hypercholesterolemia (HoFH). HeFH is the more common and less severe form of the disease, where one allele is affected and the other is normal. HoFH is characterized by the involvement of both inherited alleles, leading to greater severity that can trigger fatal cardiovascular clinical manifestations from early childhood [1,2]. Historically, according to the European Atherosclerosis Society (EAS) Consensus in 2014, the estimated prevalence of HeFH was 1:200 and consequently 1 in 160,000-300,000 for HoFH [3]. However, actual data are likely higher since both conditions are often underdiagnosed [2,3]. The overall current prevalence of familial hypercholesterolemia, including both forms, is 0.32% [4]; it is estimated that less than 5% of FH patients are diagnosed, highlighting the importance of timely genetic diagnosis [2,3].
Genetic alteration in FH can lead to the synthesis of a dysfunctional or inactive protein, decreasing cellular absorption of cholesterol from Low-Density Lipoprotein (LDL) and increasing its serum level [5]. The genes mainly affected include the Low Density Lipoprotein Receptor (LDLR) in 95%, Apolipoprotein B (APOB) in 2-5%, Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in 1%, the gene encoding the LDL Receptor Adapter Protein 1 (LDLRAP1), and Apolipoprotein E (APOE) [1,5,6].
The APOB gene, located on the short arm of chromosome 2 (2p24.1), encodes apolipoprotein B, which is crucial for the formation of chylomicrons and is the ligand for the LDL receptor. There are two isoforms of this apolipoprotein: APOB 48 and APOB 100. The APOB gene, which includes 29 exons and 28 introns, encodes APOB 100, a 4536-amino acid protein expressed in hepatocytes and enterocytes [6]. Excess APOB has been associated with an increased risk of myocardial infarction and atherosclerotic cardiovascular disease [7].
Currently, variants in the APOB gene (gene identifier: NCBI Gene:338) associated with three pathologies have been documented on genetic ontology platforms like The Human Phenotype Ontology (HPO) [8]: Homozygous Familial Hypercholesterolemia ORPHA:391665 [10], Familial Hypercholesterolemia 2 (FH2) HPO: 1440109OMIM 144010, and Familial Hypobetalipoproteinemia 1 HPO: 615558 OMIM:615558 [9].
Familial hypobetalipoproteinemia is autosomal recessively inherited, causing low levels of apoB and LDL-C cholesterol, with clinical manifestations including hepatic pathologies, fatsoluble vitamin deficiencies, intestinal malabsorption, and malnutrition [11]. Type 2 familial hypercholesterolemia, with autosomal dominant inheritance and HoFH with both autosomal dominant and autosomal recessive inheritance [1,23] lead to increased LDL-C cholesterol and an elevated risk of early cardiovascular disease [1,23]. It has been reported that around 5 to 10% of variants in the APOB gene are associated with FH [3]. Described variants in the literature include: c.10030A>G; p.(Lys3344Glu) and c.11401T>A; p.(Ser3801Thr) [1], c.10579C>T [12] Thr1558Ala P (LDLRE288K) in the European population p.Arg3527Gln [13], and in the African population rs6752026 and rs679899 [14], among others.
HoFH causes elevated LDL-C levels, leading to a high risk of atherosclerotic disease with an unfavorable cardiovascular prognosis that can result in early death. It may present asymptomatically, with cardiovascular disease being the first manifestation in some patients. Other less common clinical signs include corneal arcus, xanthomas, and xanthelasma [2,3,15-17] Lipid profile studies typically show LDL levels ≥500 mg/dl; if levels are ≥400 mg/dl, HoFH should be suspected, highlighting the importance of early diagnosis to reduce the risk of disease-associated complications [3,16]. It is essential to consider a family history of premature coronary disease and consanguinity [3].
FH diagnosis is made according to the Dutch Lipid Clinics Network Diagnostic Criteria or Simon Broome Register Criteria [14], taking into account perinatal, personal, and family history as well as paraclinical findings of elevated LDL-C levels with or without clinical signs [2,3,16]. Recent familial hypercholesterolemia guidelines from the European Atherosclerosis Society (2023) [3] emphasize early detection of hypercholesterolemia through screenings starting at age 2 in children with a family history of hypercholesterolemia or premature disease [3].
In childhood, diagnosing Familial Hypercholesterolemia (FH) can be challenging due to often nonspecific clinical manifestations; patients usually do not present a hypercholesterolemia and arterial atherosclerosis phenotype, leading to late diagnosis at an average age of 12 years [15,18]. Therefore, diagnosis through serum LDL-C measurement and family history should be complemented with molecular genetic testing [15,18].
For pathogenic variants of the LDLR, PCSK9, and APOB genes, patients are at higher risk of having elevated LDL-C levels (often 95th percentile according to country-specific age and gender criteria) and a family history of premature cardiovascular death [3,17]. Diagnostic confirmation is based on LDL levels, and ideally, genetic testing should be performed [14,18-24].
Genetic Cascade Screening (CGC) has been the main strategy for identifying patients with FH [3,20,21-24] showing effective results in the Netherlands [25]. This strategy is used to study first-degree relatives of an individual affected by FH variants by applying clinical criteria and genetic analysis. CGC is an effective method to increase the detection of patients with FH and avoid late diagnosis of the disease. If a relative tests positive for FH, testing expands to other relatives in a cascade, starting with the first degree of consanguinity and continuing successively through the rest of the family group. The goal is to establish appropriate treatment from childhood to control LDL levels and reduce the inherent cardiovascular risk in these patients [3,20,21-24].
The molecular diagnosis of FH is performed by identifying heterozygous or biallelic pathogenic variants in the LDLR, PCSK9 (gain or loss of function), APOB genes, or biallelic LDLRAP1 type genes [5,23,24]. Studies on causative variants of FH, including variants in regions of the APOB gene, have utilized genetic studies ranging from Sanger sequencing, Next-Generation Sequencing (NGS), MLPA (multiplex ligation-dependent probe amplification), to Exome Sequencing (ES) or Whole-Genome Sequencing (WGS). These diagnostic tools are useful as they allow for the identification of variants outside the usually investigated regions, contributing to new diagnostic approaches in FH [23,24,26].
Performing both individual and cascade genetic testing provides greater diagnostic certainty, facilitating timely and specific therapeutic access along with the patient’s clinical phenotype [23]. This contributes to reducing morbidity and mortality and ruling out differential diagnoses. However, it is important to consider the limitations of these tests, such as cost, possible interpretive errors, and lack of knowledge about the clinical relevance of many detected genetic variants [23-26].
The treatment of FH requires a comprehensive approach that combines lifestyle changes and pharmacological therapy, indicated if there are no significant changes in LDL-C levels or in Homozygous Familial Hypercholesterolemia (HoFH) [3]. Firstline medications are statins, which should be started between ages 6 and 10 to reduce LDL-C levels. The second line of treatment includes ezetimibe from age 10, and in severe cases, PCSK9 inhibitors can be used. In children over 10 years old, the goal is to achieve LDL-C levels below 135 mg/dL [3].
In this report, we present the clinical case of a 2-year and 4-month-old patient with no confirmed family history of genetic diseases related to lipid metabolism. During a screening study, elevated LDL levels were detected, raising suspicions of a potential genetic origin at the initial diagnosis age (6 months), with subsequent clinical and imaging findings. Based on these concerns, a genetic test was conducted to identify potential variants associated with familial hypercholesterolemia.
The genetic analysis of the patient revealed a heterozygous missense variant, c.7681C>T (p.Leu2561Phe), not previously described in the literature, with extremely low population prevalence. Based on the observed heterozygous phenotype, it is suggested that the associated pathologies could follow an autosomal dominant inheritance pattern.
Bioinformatics tools were used to deepen the genetic analysis, especially in situations where the test identifies variants of uncertain significance [27-41]. These tools employ advanced algorithms and rely on genetic databases, which allow for proper interpretation and prediction of the impact of these variants on the function of genes involved in lipid metabolism.
The results obtained facilitated comparison with other clinical cases and were integrated into new biomedical research approaches, such as reverse phenotyping, bioinformatic tools, and algorithms that predict the potential biological, molecular, and functional pathogenicity, allowing for a personalized diagnostic approach to genotype/endotype/phenotype correlation [27], thus improving diagnostic accuracy. Moreover, it enables a better understanding of genetic function, the replication of findings in animal and cellular models, and advances research into rare diseases by identifying new therapeutic targets. All of this brings us closer to personalized medicine [28,29] guiding therapeutic decisions based on the patient’s individual genetic profile.
Case report
A 2-year and 4-month-old female patient was referred to the genetics service due to abnormal laboratory results taken at 6 months of age during routine screening for growth and development, which showed the following lipid levels: Total Cholesterol (TC): 238 mg/dL (laboratory reference <200 mg/dL), Triglycerides (TG): 130 mg/dL (laboratory reference <150 mg/ dL), HDL: 44 mg/dL (laboratory reference >50 mg/dL), LDL: 216 mg/dL (laboratory reference <100 mg/dL), Thyroid-Stimulating Hormone (TSH): 2.1 μIU/mL (laboratory reference 0.7-4.5 μIU/ mL), and Estradiol: 14 pg/mL (prepubertal laboratory reference <10 pg/mL). Based on these results, the patient was initially referred to pediatric endocrinology with recommendations for healthy dietary habits.
The patient was the product of the mother’s first pregnancy, without complications during pregnancy or delivery, full-term pregnancy, delivered vaginally, with good neonatal adaptation, normal anthropometry, and non-consanguineous parents. Her family history included a maternal great-grandmother with cerebrovascular disease at age 50 and her maternal grandmother diagnosed with hypercholesterolemia and hepatic steatosis at 60 years. There were no known or confirmed other family cases of genetically inherited lipid metabolism disorders. No reports of premature death or sudden death were found in other family members. Her medical history included tonsillar hypertrophy.
Paraclinical history and medical interventions
At 1 year and 4 months: LDL: 326 mg/dL, triglycerides: 85 mg/dL, HDL: 55 mg/dL, VLDL: 17 mg/dL, and glucose: 93 mg/ dL (laboratory reference 70-100 mg/dL). At this age, nutritional modifications and treatment with Alpha-Linolenic Acid (ALA), Eicosapentaenoic Acid (EPA), and Docosahexaenoic Acid (DHA), two capsules daily, were initiated.
At 1 year and 10 months: Laboratory results showed TC: 315 mg/dL, TG: 96 mg/dL, LDL: 265 mg/dL. No additional studies were conducted.
Additional laboratory results showed apolipoprotein B: 134.5 mg/dL (laboratory reference 50-150 mg/dL), TSH: 4.59 μIU/mL (laboratory reference 0.7-4.5 μIU/mL), free T4 (T4L): 1.10 ng/ dL (laboratory reference 0.8-1.8 ng/dL), Aspartate Aminotransferase (AST): 32 U/L, Alanine Aminotransferase (ALT): 11 U/L, Erythrocyte Sedimentation Rate (ESR): 24 mm/h (laboratory reference <20 mm/h), leukocytes: 6,470 cells/μL (laboratory reference 6,000-17,500 cells/μL), C-Reactive Protein (CRP): 0.20 mg/L (laboratory reference <5 mg/L), hemoglobin: 11.1 g/dL (laboratory reference 11-13 g/dL), hematocrit: 33.8% (laboratory reference 34-40%). No other data were reported. Enzyme assays for Mucopolysaccharidosis (MPS) types I, II, IVA, VI, and VII in blood collected on filter paper showed no significant results. An abdominal ultrasound revealed hepatomegaly, with the right lobe measuring 8.6 cm, which is enlarged for her age.
On physical examination at the age of 2 years and 4 months, the patient weighed 9.20 kg and measured 76 cm, with a BMI of 16.14 kg/m². She presented generalized pallor, acanthosis nigricans on the neck folds, a non-incarcerated umbilical hernia, and mild hepatomegaly on abdominal palpation.
Based on the above findings and the high clinical, laboratory, and imaging probability of a genetic lipid metabolism disorder, a molecular study with whole exome sequencing using NextGeneration Sequencing (NGS) and Copy Number Variant (CNV) analysis was requested for genes associated with familial hypercholesterolemia (four candidate genes reported by the laboratory): APOB, LDLR, LDLRAP1, and PCSK9. Genetic counseling was provided to the father before the test.
Results
Sequencing by NGS + CNV in genes associated with familial hypercholesterolemia (four genes): APOB, LDLR, LDLRAP1, and PCSK9 was performed by the laboratory.
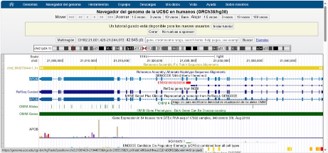
A heterozygous variant in the APOB gene (NM_000384.3) was identified, consisting of a cytosine-to-thymine change at position 7,681 (c.7681C>T) in the cDNA corresponding to exon 26/29. At the protein level, this results in a missense change from leucine to phenylalanine at codon 2,561 (p.Leu2561Phe), reference rs748303489. This variant was initially reported as having Uncertain Clinical Significance (VUS). No CNVs were identified in the analyzed genes.
Methodology and limitations
Upon further review of this variant using additional tools and algorithms, it was identified as NM_000384.3 (APOB): c.7681C>T (p.Leu2561Phe), a single-nucleotide variant. Cytogenetic location: 2p24.1 2: 21009187 (GRCh38) [NCBI UCSC] 2: 21232059 (GRCh37) [NCBI UCSC]. It is classified as a germline missense variant, Canonical SPDI: NC_000002.12:21009186:G, resulting in a protein change L2561F. It is listed in ClinVar (criteria provided, single submitter) and InterVar as a variant of uncertain significance. The allele frequency is extremely low across databases, classified with a recommended PM2 frequency threshold for the gene: 0.125%.
According to gnomAD v.4.1, exome frequency is ƒ = 0.0000103 (CoV: 69.6), and genome frequency is ƒ = 0.0000329 (CoV: 31.4). It is not reported in reference databases such as The Human Gene Mutation Database (HGMD) or the Leiden Open Variation Database (LOVD). In-silico predictors classify its significance as uncertain: BLOSUM score of 0 (BLOSUM100 version); DANN score of 0.9962; CERNIR score of 0.007; and SIFT4G score of 0.007 (dbNSFP version 4.9).
Using additional interpretation strategies, such as artificial intelligence, Var Chat reports that the genomic variant c.7681C>T is a single nucleotide substitution in the APOB gene, resulting in the amino acid change from leucine to phenylalanine at position 2561 (p.Leu2561Phe) in the apolipoprotein B protein. This variant is cataloged in the dbSNP database with reference SNP ID rs748303489. The c.7681C>T variant results in a non-conservative amino acid change in the apoB-100 isoform, potentially affecting the protein’s structure and function. Given the significance of apoB-100 in lipid transport and metabolism, alterations in its structure could disrupt normal lipoprotein formation and function, leading to dyslipidemia and related disorders. Variants in the APOB gene account for a notable percentage of FH cases, though they are less common than those in the LDLR gene. The c.7681C>T (p.Leu2561Phe) variant in this case has not been previously described, underscoring the importance of reporting new variants and utilizing methods like reverse phenotyping to clarify their clinical implications. The absence of other known variants and the persistence of elevated LDL levels suggest that other genetic or environmental factors may contribute to the patient’s hypercholesterolemia.
According to Genome Explorer AI, the potential impact is as follows: Leucine (Leu) is a hydrophobic amino acid, while phenylalanine (Phe) is also hydrophobic but has a bulkier aromatic side chain. This substitution may impact protein structure or function, depending on where it occurs in the apolipoprotein B protein. Variants in APOB are associated with conditions like familial hypercholesterolemia, a genetic disorder leading to high cholesterol levels and increased cardiovascular risk. The clinical significance of this variant would depend on additional information, such as population frequency, functional studies, or clinical presentation.
According to UniProt, apolipoprotein B is a major constituent of chylomicrons (apo B-48), LDL (apo B-100), and VLDL (apo B-100), with apo B-100 serving as a recognition signal for cellular binding and internalization of LDL particles via the apoB/E receptor.
According to BIOGPS, the APOB gene has accessions: 338 (NCBI Gene), ENSG00000084674 (Ensembl), ENSG00000291544 (Ensembl), P04114 (UniProt), 107730 (OMIM), and 328 (HomoloGene). Molecular Function includes protein binding (GO:0005515), phospholipid binding (GO:0005543), heparin binding (GO:0008201), lipase binding (GO:0035473), receptor ligand activity (GO:0048018), low-density lipoprotein particle receptor binding (GO:0050750), cholesterol transfer activity (GO:0120020). Biological Process involves in utero embryonic development (GO:0001701), triglyceride mobilization (GO:0006642), signal transduction (GO:0007165), spermatogenesis (GO:0007283), nervous system development (GO:0007399), cholesterol metabolic process (GO:0008203), and more. ApoB interacts with apo(a), PPIB, the Calcitonin receptor, and HSP90B1, and its interaction with proteoglycans, collagen, and fibronectin is believed to cause atherosclerosis.
Currently, variants in the APOB gene (gene identifier: NCBI Gene:338) associated with three pathologies have been documented on genetic ontology platforms like The Human Phenotype Ontology (HPO): Homozygous Familial Hypercholesterolemia ORPHA:39166510, Familial Hypercholesterolemia 2 (FH2) HPO: 1440109OMIM 144010, and Familial Hypobetalipoproteinemia 1 HPO: 615558 OMIM:615558.
In light of the above findings, current methodological and interpretative limitations, and the importance of reverse phenotyping-through a multimodal and transdisciplinary assessment in such diagnostically challenging cases, and given the characteristics of the patient (diagnosis at 6 months of age; persistent abnormal clinical, laboratory, and imaging findings despite age-appropriate therapeutic options; altered imaging studies; absence of confirmed family diagnosis or genetic studies; with a genetic result showing a variant in the APOB gene associated with autosomal dominant inheritance according to related ontological platforms like HPO, OMIM, Gene Ontology (GO), ORPHANET), further examination of this variant using computational tools and algorithms suggests multiple lines of computational evidence supporting a deleterious effect on the gene or gene product. This variant results in a non-conservative amino acid change in the apoB-100 isoform, which may impact protein structure and function. It is not reported in reference databases such as NCBI, MedGen, HGMD, OMIM, the 1000 Genomes Project, ExAc, LOVD3, Ensembl, ClinGen, or DGV. However, in-silico predictive algorithms (BLOSUM, DANN, CERNIR, SIFT4G) have reclassified its significance from benign to uncertain. Several platforms with biological, genomic annotations, molecular bases, and protein structure and function databases (BioGPS, UniProt), as well as artificial intelligence tools (VarChat, Genome Explorer AI), predict a deleterious effect. According to Richards et al., Standards and Guidelines for the Interpretation of Sequence Variants, 2015, American College of Medical Genetics and Genomics, Association for Molecular Pathology, ClinGen, and using pipeline algorithms, this variant is classified with evidence: PM2, PM6, PP3; suggesting that from a holistic, integrated, and personalized approach, it should be considered likely pathogenic, de novo occurrence with extremely low population prevalence.
Discussion
Familial Hypercholesterolemia (FH) is associated with elevated LDL cholesterol levels from an early age, involving genes such as LDLR, LDLRAP1, PCSK9, APOE, and APOB [1,5,6]. Pathogenic variants in the APOB gene play a significant role in FH [30], as this gene is crucial for LDL particle binding and catabolism, contributing to the various phenotypes of FH and serving as a key marker in assessing atherosclerosis and cardiovascular risk [5,31]. Identifying genetic variants in these genes may offer more precision than direct LDL measurements [3,1].
The genetic criteria defined by the European Atherosclerosis Society (EAS) are based on identifying pathogenic or likely pathogenic biallelic variants in the LDLR, APOB, PCSK9, or LDLRAP1 genes, or detecting at least two of these variants in different loci [3].
In our clinical case, we describe a patient with persistently abnormal LDL results from six months of age, despite dietary adjustments and age-appropriate pharmacological treatment, later accompanied by clinical and imaging abnormalities. The patient had no known or confirmed family history of genetic lipid metabolism disorders. Genetic analysis identified a variant in the APOB gene, initially classified as a Variant of Uncertain Significance (VUS). With the development of multimodal resources and interpretative tools, including artificial intelligence, we highlight the importance of a personalized evaluation for potential phenotype-endotype-genotype correlation.
It is essential to complement this with other strategies, such as segregation studies and cascade screening, which is invaluable for identifying other affected family members. In our patient’s case, genetic counseling recommended diagnostic testing for first and second-degree relatives who had not yet undergone cardiovascular risk evaluation [3,20-24].
Among the variants reported in the literature, recent studies, such as that by Li Bobby et al. in 2024 [32], conducted a case control study in New Zealand adults, including 213 patients with positive genetic panels for LDLR, APOB, PCSK9, and APOE genes. Of these, 14.6% had pathogenic or likely pathogenic FH variants, and 4.7% had variants of uncertain significance [32].
In 2023, Kammal et al. sequenced three patients from a database of 1000 genomes, identifying the p.Arg3527Trp variant in the APOB gene in one patient. This variant was classified as pathogenic because it replaces arginine with tryptophan, altering protein folding and stability due to its critical position in the APOB gene [15].
In Qingdao, China, Zhou Y et al. reported in 2024 [33] the results of whole-genome sequencing of 6,820 newborns, identifying FH variants in the LDLR, APOB, and PCSK9 genes, with 35 heterozygous carriers of FH variants and no homozygous or compound heterozygous cases. Notable variants included LDLR c.1747C>T, LDLR c.1448G>A, and APOB c.10579C>T, with an FH prevalence of 0.47% (95% CI: 0.32%-0.66%). Two novel mutations, LDLR (c.1592T>A p.Met531Lys) and PCSK9 (c.706G>A p.Gly236Ser), were reported as variants of uncertain significance [33].
At the Children’s Hospital in Turin, Italy, Buganza et al. (2024) [14] examined 180 pediatric patients at the Lipid Clinic with an average age of 10.4±4.6 years. Among these, 64 patients were diagnosed with FH. Mutations in LDLR were identified in most subjects, with only two cases in the APOB and PCSK9 genes. The overall mutation detection rate was 91.1% [14].
In 2024, Cardozo Gasparin et al. [34] investigated APOE and APOB polymorphisms in adults of European ancestry from southern Brazil, studying the association between the R3500Q mutation in the APOB gene and dyslipidemia. The R3500Q variant has been linked to Familial Defective Apolipoprotein B-100 (FDB) [34]. They noted that the ε4 allele of APOE was more frequent in individuals with high total cholesterol and LDL-C levels, whereas the ε2 allele was more frequent in individuals with high HDL-C, indicating a protective effect [34].
The APOB gene contributes to 2-5% of FH cases [5,6]. Rodríguez-Jiménez C et al. (2023) [35] found that among 825 hypercholesterolemic patients, 40% had a variant in LDLR, APOB, PCSK9, or LDLRAP1. Of these, 12% had APOB variants [35]. The p.(Arg3527Gln) variant was most frequently observed, with other variants such as c.10030A>G and p.(Lys3344Glu) recently reported [35].
In the Amish community, the APOB variant c.10580G>A (p.Arg3527Gln) has a minor allele frequency of 6.7%, compared to 0.08% in European populations. Heterozygous carriers of this variant have been associated with elevated LDL-C and coronary artery calcification, emphasizing the value of genetic screening [36,37].
In Japanese patients, Chaudhry A et al. (2023) [38] reported the APOB variant p.(Pro955Ser) with an allele frequency of 0.15 in heterozygous FH (HeFH) patients, significantly higher than in the general Japanese population (0.034), with an odds ratio of 4.9. This variant may moderately influence FH severity, with age, diet, and environmental factors affecting its expression [38].
Other polymorphisms in APOB, such as rs5742904 (R3500Q), have been linked to LDL-C levels and coronary calcification [38]. The absence of this polymorphism has been associated with better cardiovascular health in studies of longevity [39].
The updated EAS criteria for diagnosing HoFH include measuring untreated plasma LDL-C levels >10 mmol/L (>∼400 mg/ dL), with additional clinical findings like xanthomas before age 10 [3]. Recent findings by Li BV et al. (2024) [32] and Kamal A et al. (2023) [15] highlight the importance of early clinical manifestations, such as tendon xanthomas and corneal arcus, in diagnosing HoFH.
Aparicio A et al. (2023) described pathogenic variants in LDLR, APOB, PCSK9, and LDLRAP1 in 182 Spanish patients, noting that the average age of diagnosis was between 25 and 35 years, with genetic confirmation around 42 to 54 years [16].
In our patient, physical examination revealed no xanthomas, xanthelasmas, or corneal arcus abnormalities. However, a nonincarcerated umbilical hernia, mild hepatomegaly, and acanthosis nigricans were noted at age two, with LDL levels reaching a maximum of 326 mg/dL, consistent with reports in the literature [3].
FH treatment combines lifestyle modifications and pharmacological therapy, with statins recommended for children aged 6 to 10 to reduce LDL-C levels [3]. In our case, initial treatment involved dietary changes and omega-3 supplementation. Since the patient was under six, other therapies were not initiated due to age-related restrictions.
Timely genetic diagnosis is crucial to mitigate future cardiovascular risks, such as premature atherosclerosis. Identifying affected children early enables therapeutic strategies, including lifestyle changes and statins, which improve long-term outcomes. Genetics is essential for diagnosis, treatment guidance, and family counseling [3].
Integrating new evidence is vital for interpreting variants, especially those not characterized in databases. A multimodal approach, including bioinformatics, enhances the identification of variants of uncertain significance [39,40].
This multidisciplinary approach advances personalized medicine, allowing therapeutic decisions based on individual genetic profiles and enabling targeted treatments that improve outcomes for hereditary diseases like FH [39,40].
Conclusion
FH is a significant genetic disease in pediatrics, associated with high morbidity and mortality due to cardiovascular complications from prolonged exposure to elevated LDL-C levels. Early detection is essential, as underdiagnosis is common when based solely on clinical criteria. Cascade genetic screening significantly impacts the pediatric population by enabling early intervention to reduce the disease burden.
Inverse phenotyping is a valuable diagnostic strategy, transforming clinical significance from uncertain to pathogenic through detailed phenotype analysis. Multimodal tools, including computational systems, enable personalized diagnoses. Advanced genetic studies, such as exome or whole-genome sequencing, offer accurate diagnoses.
Continued characterization of genetic variants in FH is necessary to confirm pathogenicity and improve diagnostic and therapeutic approaches within precision medicine. Early pharmacological interventions reduce cardiovascular risk, enhancing quality of life and prognosis. Diagnostic approaches, such as inverse phenotyping and computational tools, reclassify uncertain variants, leading to precise, personalized diagnoses. Despite current methodological and interpretative limitations, personalized, targeted, and transdisciplinary assessments are essential for contemporary medical challenges. Early diagnoses support targeted treatments, genetic counseling, segregation studies, and monitoring, bringing us closer to 7P Medicine personalized, predictive, preventive, participative, proactive, precision, and population-based.
References
- Van den Bosch SE, Corpeleijn WE, Hutten BA, Wiegman A. How Genetic Variants in Children with Familial Hypercholesterolemia Not Only Guide Detection, but Also Treatment. Genes (Basel). 2023; 14(3): 669. doi: 10.3390/genes14030669.
- Timoshchenko O, Ivanoshchuk D, Semaev S, Orlov P, Zorina V, et al. Diagnosis of Familial Hypercholesterolemia in Children and Young Adults. Int J Mol Sci. 2023; 25(1): 314. doi: 10.3390/ ijms25010314.
- Cuchel M, Raal FJ, Hegele RA, Al-Rasadi K, Arca M, et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: New treatments and clinical guidance. Eur Heart J. 2023; 44(25): 2277-2291. doi: 10.1093/eurheartj/ehad197.
- Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J Am Coll Cardiol. 2020; 75(20): 2553-2566. doi: 10.1016/j.jacc.2020.03.057.
- Abifadel M, Boileau C. Genetic and molecular architecture of familial hypercholesterolemia. J Intern Med. 2023; 293(2): 144-165. doi: 10.1111/joim.13577.
- Borén J, Taskinen MR, Packard CJ. Biosynthesis and Metabolism of ApoB-Containing Lipoproteins. Annu Rev Nutr. 2024; 44(1): 179-204. doi: 10.1146/annurev-nutr-062222-020716.
- Remaley AT, Cole J, Sniderman AD. ApoB Is Ready for Prime Time. J Am Coll Cardiol. 2024; 83(23): 2274-2275. doi: 10.1016/j. jacc.2024.04.008. PMID: 38839201.
- Gargano MA, Matentzoglu N, Coleman B, Addo-Lartey EB, Anagnostopoulos AV, et al. The Human Phenotype Ontology in phenotypes around the world. Nucleic Acids Res. 2024; 52(D1): D1333-D1346. doi: 10.1093/nar/gkad1005.
- Human Phenotype Ontology Consortium. Human Phenotype Ontology. NCBIGene: 338. Recuperado de. 2024. https://hpo. jax.org/browse/gene/NCBIGene:338
- Orphanet. Orphanet: Homozygous familial hypercholesterolemia. Recuperado de. 2024. https://www.orpha.net/en/dis- ease/detail/391665
- Burnett JR, Hooper AJ, Hegele RA. APOB-Related Familial Hypobetalipoproteinemia. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle. 2021; 1993-2024.
- Hori M, Takahashi A, Hosoda K, Ogura M, Harada-Shiba M. Una variante de APOB p.(Pro955Ser) de baja frecuencia contribuye a la gravedad y variabilidad de la hipercolesterolemia familiar. J Clin Endocrinol Metab. 2023; 108(2): 422-432.
- Y Luo G, Zhang A, Gao S, Tang Y, Du Z, et al. Genetic identification of familial hypercholesterolemia within whole genome sequences in 6820 newborns. Clin Genet. 2024; 105(3): 308-312. doi: 10.1111/cge.14453.
- Buganza R, Massini G, Di Taranto, Cardiero G, de Sanctis L, et al. Simplified Criteria for Identification of Familial Hypercholesterolemia in Children: Application in Real Life. J Cardiovasc Dev Dis. 2024; 11(4): 123. doi: 10.3390/jcdd11040123.
- Kamal A, Kanchau JD, Shahuri NS, Mohamed-Yassin MS, Baharudin N, et al. Case Series of Genetically Confirmed Index Cases of Familial Hypercholesterolemia in Primary Care. Am J Case Rep. 2023; 24: e939489. doi: 10.12659/AJCR.939489.
- Aparicio A, Villazón F, Suárez-Gutiérrez L, Gómez J, MartínezFaedo C, et al. Evaluación clínica de pacientes con hipercolesterolemia familiar genéticamente confirmada. J Clin Med. 2023; 12(3): 1030. doi: 10.3390/jcm12031030.
- Alves AC, Alonso R, Diaz-Diaz JL, Medeiros AM, Jannes CE, et al. Phenotypical, Clinical, and Molecular Aspects of Adults and Children with Homozygous Familial Hypercholesterolemia in Iberoamerica. Arterioscler Thromb Vasc Biol. 2020; 40(10): 2508-2515. doi: 10.1161/ATVBAHA.120.313722.
- Harada-Shiba M, Ohtake A, Sugiyama D, Tada H, Dobashi K, et al. Guidelines for the Diagnosis and Treatment of Pediatric Familial Hypercholesterolemia 2022. J Atheroscler Thromb. 2023; 30(5): 531-557. doi: 10.5551/jat.CR006.
- Lokkesmoe R, Hamilton L. The Role of Reverse Cascade Screening in Children with Familial Hypercholesterolemia: A Literature Review and Analysis. Curr Atheroscler Rep. 2024; 26(8): 427-433. doi: 10.1007/s11883-024-01211-9.
- Khoury M. Cascade Screening in Familial Hypercholesterolemia: Achieving Buy-In and Turning Patients into Partners. CJC Pediatr Congenit Heart Dis. 2023; 2(5): 219-221. doi: 10.1016/j.
- Yip MK, Kwan EY, Leung JY, Lau EY, Poon WT. Genetic Spectrum and Cascade Screening of Familial Hypercholesterolemia in Routine Clinical Setting in Hong Kong. Genes (Basel). 2023; 14(11): 2071. doi: 10.3390/genes14112071.
- Watts GF, Gidding SS, Hegele RA, et al. International Atherosclerosis Society guidance for implementing best practice in the care of familial hypercholesterolaemia. Nat Rev Cardiol. 2023; 20(12): 845. doi: 10.1038/s41569-023-00892-0.
- Dickson MA, Zahavich L, Rush J, et al. Exploring barriers and facilitators to indirect cascade screening for familial hypercholesteraemia in a paediatric/parent population. CJC Pediatr Congenit Heart Dis. 2023; 2(5): 211. doi: 10.1016/j.cjcpc.2023.05.006.
- Ontario Health (Quality). Genetic Testing for Familial Hypercholesterolemia: Health Technology Assessment. Ont Health Technol Assess Ser. 2022; 22(3): 1-155.
- Elbitar S, Susan-Resiga D, Ghaleb Y, El Khoury P, Peloso G, et al. New Sequencing technologies help revealing unexpected mutations in Autosomal Dominant Hypercholesterolemia. Sci Rep. 2018; 8(1): 1943. doi: 10.1038/s41598-018-20281-9.
- Sturm AC, Knowles JW, Gidding SS, Ahmad ZS, Ahmed CD, et al. Convened by the Familial Hypercholesterolemia Foundation. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2018; 72(6): 662-680. doi: 10.1016/j.jacc.2018.05.044.
- Wilczewski CM, Obasohan J, Paschall JE, Zhang S, Singh S, et al. Genotype first: Clinical genomics research through a reverse phenotyping approach. Am J Hum Genet. 2023; 110(1): 3-12. doi: 10.1016/j.ajhg.2022.12.004.
- Franks PW, Cefalu WT, Dennis J, Florez JC, Mathieu C, et al. Precision medicine for cardiometabolic disease: A framework for clinical translation. Lancet Diabetes Endocrinol. 2023; 11(11): 822-835. doi: 10.1016/S2213-8587(23)00165-1.
- Tesi B, Boileau C, Boycott KM, Canaud G, Caulfield M, et al. Lindstrand A. Precision medicine in rare diseases: What is next? J Intern Med. 2023; 294(4): 397-412. doi: 10.1111/joim.13655.
- Medeiros AM, Bourbon M. Genetic Testing in Familial Hypercholesterolemia: Is It for Everyone? Curr Atheroscler Rep. 2023; 25(4): 127-132. doi: 10.1007/s11883-023-01091-5.
- Jigoranu RA, Roca M, Costache AD, Mitu O, Oancea AF, et al. Novel Biomarkers for Atherosclerotic Disease: Advances in Cardiovascular Risk Assessment. Life (Basel). 2023; 13(8): 1639. doi: 10.3390/life13081639.
- Li BV, Laurie AD, Reid NJ, Leath MA, King RI, et al. Association of Clinical Characteristics With Familial Hypercholesterolaemia Variants in a Lipid Clinic Setting: A Case-Control Study. J Lipid Atheroscler. 2024; 13(1): 29-40. doi: 10.12997/jla.2024.13.1.29.
- Zhou Y, Luo G, Zhang A, Gao S, Tang Y, et al. Genetic identification of familial hypercholesterolemia within whole genome sequences in 6820 newborns. Clin Genet. 2024; 105(3): 308-312. doi: 10.1111/cge.14453.
- Cardozo Gasparin, C, Viater Tureck L, Pereira Ferrari L, Lehtonen Rodrigues de Souz R, Furtado Alle L. Polymorphisms of APOE and APOB genes and dyslipidemias in a South Brazilian Cohort. Brazilian Journal of Implantology and Health Sciences. 2024; 6(1): 992-1012. https://doi.org/10.36557/26748169.2024v6n1p992-1012.
- Rodríguez-Jiménez C, de la Peña G, Sanguino J, Poyatos-Peláez S, Carazo A, et al. Rodríguez-Nóvoa S. Identification and Functional Analysis of APOB Variants in a Cohort of Hypercholesterolemic Patients. Int J Mol Sci. 2023; 24(8): 7635. doi: 10.3390/ ijms24087635.
- Williams KB, Horst M, Young M, Pascua C, Puffenberger EG, et al. Clinical characterization of familial hypercholesterolemia due to an amish founder mutation in Apolipoprotein B. BMC Cardiovasc Disord. 2022; 22(1): 109. doi: 10.1186/s12872-022-02539-3.
- Snyder C, Beitelshees AL, Chowdhury D. Familial Hyperlipidemia Caused by Apolipoprotein B Mutation in the Pediatric Amish Population: A Mini Review. Interv Cardiol (Lond). 2023; 15(17): 433-437.
- Chaudhry A, Trinder M, Vesely K, Cermakova L, Jackson L, et al. Genetic Identification of Homozygous Familial Hypercholesterolemia by Long-Read Sequencing Among Patients With Clinically Diagnosed Heterozygous Familial Hypercholesterolemia. Circ Genom Precis Med. 2023; 16(2): e003887. doi: 10.1161/CIRCGEN.122.003887.
- Kondakova EV, Ilina VM, Ermakova LM, Krivonosov MI, Kuchin KV, et al. New Genetically Determined Markers of the Functional State of the Cardiovascular System. Genes (Basel). 2023; 14(1): 185. doi: 10.3390/genes14010185.
- Guerin A, Iatan I, Ruel I, Ngufor LF, Genest J. Genetic testing for familial hypercholesterolemia in Quebec, Canada: A single-centre retrospective cohort study. CMAJ Open. 2023; 11(4): E754E764.
- Larrea-Sebal A, Jebari-Benslaiman S, Galicia-Garcia U, JoseUrteaga AS, Uribe KB, et al. Predictive Modeling and Structure Analysis of Genetic Variants in Familial Hypercholesterolemia: Implications for Diagnosis and Protein Interaction Studies. Curr Atheroscler Rep. 2023; 25(11): 839-859. doi: 10.1007/s11883-023-01154-7.